Our Product Portfolio
The following table summarizes our portfolio:
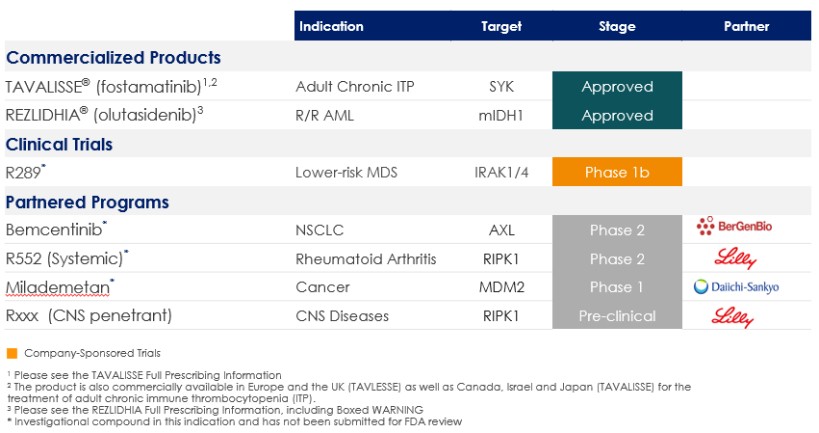
Commercial Products
TAVALISSE/Fostamatinib in ITP
Chronic ITP affects an estimated 81,300 adult patients in the US. In patients with ITP, the immune system attacks and destroys the body’s own blood platelets, which play an active role in blood clotting and healing. ITP patients can suffer extraordinary bruising, bleeding and fatigue as a result of low platelet counts. Current therapies for ITP include steroids, blood platelet production boosters that imitate thrombopoietin (TPO) and splenectomy.
Taken in tablet form, fostamatinib blocks the activation of SYK inside immune cells. ITP is typically characterized by the body producing antibodies that attach to healthy platelets in the blood stream. Immune cells recognize these antibodies and affix to them, which activates the SYK enzyme inside the immune cell, and triggers the destruction of the antibody and the attached platelet. When SYK is inhibited by fostamatinib, it interrupts this immune cell function and allows the platelets to escape destruction. The results of our Phase 2 clinical trial, in which fostamatinib was orally administered to 16 adults with chronic ITP, published in Blood, showed that fostamatinib significantly increased the platelet counts of certain ITP patients, including those who had failed other currently available agents.
Our Fostamatinib for Immune Thrombocytopenia (FIT) Phase 3 clinical program had a total of 150 ITP patients which were randomized into two identical multi-center, double-blind, placebo-controlled clinical trials. The patients were diagnosed with persistent or chronic ITP, and had blood platelet counts consistently below 30,000 per microliter of blood. Two-thirds of the subjects received fostamatinib orally at 100 mg twice daily (bid) and the other third received placebo on the same schedule. Subjects were expected to remain on treatment for up to 24 weeks. At week four of treatment, subjects who failed to meet certain platelet counts and met certain tolerability thresholds could have their dosage of fostamatinib (or corresponding placebo) increased to 150 mg bid. The primary efficacy endpoint of this program was a stable platelet response by week 24 with platelet counts at or above 50,000 per microliter of blood for at least four of the final six qualifying blood draws. In August 2016, we announced the results of the first FIT study, reporting that fostamatinib met the study’s primary efficacy endpoint. The study showed that 18% of patients receiving fostamatinib achieved a stable platelet response compared to none receiving a placebo control. In October 2016, we announced the results of the second FIT study, reporting that the response rate (16% in the treatment group, versus 4% in the placebo group) was consistent with the first study, although the difference was not statistically significant. In the ITP double-blind studies, the most commonly reported adverse reactions occurring in at least 5% of patients treated with TAVALISSE were diarrhea, hypertension, nausea, dizziness, increased alanine aminotransferase, increased aspartate
29