UNITED STATES
SECURITIES AND EXCHANGE COMMISSION
WASHINGTON, D.C. 20549
FORM 10-Q
|
ý |
QUARTERLY REPORT PURSUANT TO SECTION 13 OR 15(d) OF THE SECURITIES EXCHANGE ACT OF 1934 |
FOR THE QUARTERLY PERIOD ENDED SEPTEMBER 30, 2002.
OR
|
o |
TRANSITION REPORT PURSUANT TO SECTION 13 OR 15(d) OF THE SECURITIES EXCHANGE ACT OF 1934 |
FOR THE TRANSITION PERIOD FROM TO .
Commission File Number 0-29889
Rigel Pharmaceuticals, Inc.
(Exact name of registrant as specified in its charter)
|
Delaware |
|
94-3248524 |
|
(State or other jurisdiction of incorporation or organization) |
|
(I.R.S. Employer Identification No.) |
|
|
|
|
|
240 East Grand Avenue |
|
94080 |
|
(Address of principal executive offices) |
|
(Zip Code) |
|
|
|
|
|
(650) 624-1100 |
||
|
(Registrant’s telephone number, including area code) |
||
Indicate by check mark whether the registrant: (1) has filed all reports required to be filed by Section 13 or 15(d) of the Securities Exchange Act of 1934 during the preceding 12 months (or for such shorter period that the Registrant was required to file such reports), and (2) has been subject to such filing requirements for the past 90 days. Yes ý No o
As of November 1, 2002, there were 45,599,102 shares of the Registrant’s common stock outstanding.
RIGEL PHARMACEUTICALS, INC.
QUARTERLY REPORT ON FORM 10-Q
FOR THE QUARTERLY PERIOD ENDED SEPTEMBER 30, 2002
INDEX
2
PART I FINANCIAL INFORMATION
Item 1. Financial Statements
RIGEL
PHARMACEUTICALS, INC.
CONDENSED BALANCE SHEETS
(in thousands, except share and per share amounts)
|
|
|
September 30, |
|
December 31, |
|
||
|
|
|
(unaudited) |
|
(Note 1) |
|
||
|
Assets |
|
|
|
|
|
||
|
Current assets: |
|
|
|
|
|
||
|
Cash and cash equivalents |
|
$ |
36,268 |
|
$ |
11,488 |
|
|
Available-for-sale securities |
|
250 |
|
21,927 |
|
||
|
Accounts receivable |
|
107 |
|
1,153 |
|
||
|
Prepaid expenses and other current assets |
|
2,728 |
|
1,965 |
|
||
|
Total current assets |
|
39,353 |
|
36,533 |
|
||
|
|
|
|
|
|
|
||
|
Property and equipment, net |
|
12,664 |
|
8,440 |
|
||
|
Other assets |
|
1,814 |
|
1,475 |
|
||
|
|
|
$ |
53,831 |
|
$ |
46,448 |
|
|
|
|
|
|
|
|
||
|
Liabilities and stockholders’ equity |
|
|
|
|
|
||
|
Current liabilities: |
|
|
|
|
|
||
|
Accounts payable |
|
$ |
5,021 |
|
$ |
1,952 |
|
|
Accrued compensation |
|
941 |
|
671 |
|
||
|
Accrued liabilities |
|
2,766 |
|
1,104 |
|
||
|
Deferred revenue |
|
4,027 |
|
3,264 |
|
||
|
Capital lease obligations |
|
3,706 |
|
3,171 |
|
||
|
Total current liabilities |
|
16,461 |
|
10,162 |
|
||
|
|
|
|
|
|
|
||
|
Capital lease obligations |
|
2,986 |
|
4,243 |
|
||
|
Long-term portion of deferred revenue |
|
1,550 |
|
2,240 |
|
||
|
Other long-term liabilities |
|
216 |
|
862 |
|
||
|
|
|
|
|
|
|
||
|
Commitments |
|
|
|
|
|
||
|
|
|
|
|
|
|
||
|
Stockholders’ equity: |
|
|
|
|
|
||
|
Common stock, $0.001 par value; 100,000,000 shares authorized; 45,544,602 and 37,732,209 shares issued and outstanding on September 30, 2002 and December 31, 2001, respectively |
|
46 |
|
38 |
|
||
|
Additional paid-in capital |
|
140,386 |
|
109,095 |
|
||
|
Deferred stock compensation |
|
(1,070 |
) |
(2,452 |
) |
||
|
Accumulated other comprehensive income |
|
— |
|
44 |
|
||
|
Accumulated deficit |
|
(106,744 |
) |
(77,784 |
) |
||
|
Total stockholders’ equity |
|
32,618 |
|
28,941 |
|
||
|
|
|
$ |
53,831 |
|
$ |
46,448 |
|
Note (1) The balance sheet at December 31, 2001 has been derived from the audited financial statements at that date included in the Company’s Form 10-K for the fiscal year ended December 31, 2001.
See accompanying notes.
3
RIGEL
PHARMACEUTICALS, INC.
CONDENSED STATEMENTS OF OPERATIONS
(in thousands, except per share amounts)
|
|
|
Three Months Ended |
|
Nine Months Ended |
|
||||||||
|
|
|
2002 |
|
2001 |
|
2002 |
|
2001 |
|
||||
|
|
|
(unaudited) |
|
(unaudited) |
|
||||||||
|
Revenues: |
|
|
|
|
|
|
|
|
|
||||
|
Contract revenues from collaborations |
|
$ |
3,653 |
|
$ |
4,206 |
|
$ |
12,088 |
|
$ |
10,524 |
|
|
|
|
|
|
|
|
|
|
|
|
||||
|
Costs and expenses: |
|
|
|
|
|
|
|
|
|
||||
|
Research and development |
|
11,624 |
|
8,631 |
|
33,781 |
|
23,395 |
|
||||
|
General and administrative |
|
2,130 |
|
1,991 |
|
7,335 |
|
5,885 |
|
||||
|
|
|
13,754 |
|
10,622 |
|
41,116 |
|
29,280 |
|
||||
|
Loss from operations |
|
(10,101 |
) |
(6,416 |
) |
(29,028 |
) |
(18,756 |
) |
||||
|
Interest income |
|
172 |
|
437 |
|
732 |
|
1,662 |
|
||||
|
Interest expense |
|
(213 |
) |
(240 |
) |
(664 |
) |
(600 |
) |
||||
|
Net loss |
|
$ |
(10,142 |
) |
$ |
(6,219 |
) |
$ |
(28,960 |
) |
$ |
(17,694 |
) |
|
Net loss per share, basic and diluted |
|
$ |
(0.22 |
) |
$ |
(0.17 |
) |
$ |
(0.65 |
) |
$ |
(0.48 |
) |
|
Weighted average shares used in computing net loss per common share, basic and diluted |
|
45,515 |
|
37,516 |
|
44,735 |
|
37,173 |
|
||||
See accompanying notes.
4
RIGEL
PHARMACEUTICALS, INC.
CONDENSED STATEMENTS OF CASH FLOWS
(in thousands)
|
|
|
Nine Months Ended |
|
||||
|
|
|
2002 |
|
2001 |
|
||
|
|
|
(unaudited) |
|
||||
|
Operating activities: |
|
|
|
|
|
||
|
Net loss |
|
$ |
(28,960 |
) |
$ |
(17,694 |
) |
|
Adjustments to reconcile net loss to net cash used in operating activities: |
|
|
|
|
|
||
|
Depreciation and amortization |
|
3,671 |
|
2,908 |
|
||
|
Amortization of deferred stock compensation |
|
777 |
|
2,119 |
|
||
|
Noncash stock compensation recovery |
|
(113 |
) |
(496 |
) |
||
|
Issuances of equity instruments for noncash benefits |
|
15 |
|
— |
|
||
|
Changes in assets and liabilities: |
|
|
|
|
|
||
|
Accounts receivable |
|
1,046 |
|
(3,013 |
) |
||
|
Prepaid expenses and other current assets |
|
697 |
|
(334 |
) |
||
|
Other assets |
|
(36 |
) |
(531 |
) |
||
|
Accounts payable |
|
1,608 |
|
(569 |
) |
||
|
Accrued compensation |
|
270 |
|
84 |
|
||
|
Accrued liabilities |
|
1,662 |
|
3 |
|
||
|
Deferred revenue |
|
73 |
|
4,475 |
|
||
|
Other long-term liabilities |
|
(646 |
) |
43 |
|
||
|
Net cash used in operating activities |
|
(19,936 |
) |
(13,005 |
) |
||
|
|
|
|
|
|
|
||
|
Investing activities: |
|
|
|
|
|
||
|
Purchase of available-for-sale securities |
|
(25,956 |
) |
(40,415 |
) |
||
|
Maturities of available-for-sale securities |
|
22,625 |
|
18,240 |
|
||
|
Sale of available-for-sale securities |
|
24,964 |
|
— |
|
||
|
Capital expenditures |
|
(7,895 |
) |
(2,623 |
) |
||
|
Net cash provided by (used in) investing activities |
|
13,738 |
|
(24,798 |
) |
||
|
|
|
|
|
|
|
||
|
Financing activities: |
|
|
|
|
|
||
|
Proceeds from capital lease financing |
|
1,999 |
|
1,748 |
|
||
|
Principal payments on capital lease obligations |
|
(2,721 |
) |
(2,470 |
) |
||
|
Net proceeds from issuances of common stock |
|
31,700 |
|
601 |
|
||
|
Net cash provided by (used in) financing activities |
|
30,978 |
|
(121 |
) |
||
|
|
|
|
|
|
|
||
|
Net increase (decrease) in cash and cash equivalents |
|
24,780 |
|
(37,924 |
) |
||
|
Cash and cash equivalents at beginning of period |
|
11,488 |
|
49,030 |
|
||
|
Cash and cash equivalents at end of period |
|
$ |
36,268 |
|
$ |
11,106 |
|
See accompanying notes.
5
Rigel Pharmaceuticals, Inc.
Notes to Condensed Financial Statements
(unaudited)
1. Nature of operations
Rigel Pharmaceuticals, Inc. (“Rigel” or the “Company”) was incorporated in the state of Delaware on June 14, 1996. The Company is engaged in the discovery and development of a broad range of new small molecule drug candidates.
2. Basis of presentation
The accompanying unaudited condensed financial statements of the Company have been prepared in accordance with generally accepted accounting principles for interim financial information and pursuant to the instructions to Form 10-Q and Article 10 of Regulation S-X. In the opinion of Rigel’s management, these unaudited condensed financial statements include all adjustments, consisting only of normal recurring adjustments, which Rigel considers necessary to fairly state the Company’s financial position and the results of its operations and its cash flows. Interim-period results are not necessarily indicative of results of operations or cash flows for a full-year period. The balance sheet at December 31, 2001 has been derived from audited financial statements at that date, but does not include all disclosures required by generally accepted accounting principles for complete financial statements.
These condensed financial statements and the notes accompanying them should be read in conjunction with the Company’s Annual Report on Form 10-K for the year ended December 31, 2001. Stockholders are encouraged to review the Form 10-K for a broader discussion of the Company’s business and the opportunities and risks inherent in the Company’s business. Copies of the Form 10-K are available from the Company upon request.
Comprehensive loss did not differ materially from the net loss as reported.
3. Net loss per share
Basic earnings per share excludes any dilutive effects of options or warrants. The calculation of diluted net loss per share excludes shares of potential common stock if the effect is anti-dilutive.
4. Revenue recognition
Non-refundable, up-front payments received in connection with research and development collaboration agreements, including technology access fees, are deferred and recognized on a straight-line basis over the relevant periods of continuing involvement, generally the research term.
Revenues related to collaborative research with the Company’s corporate collaborators are recognized as research services are performed over the related funding periods for each contract. Under these agreements, the Company is required to perform research and development activities as specified in each respective agreement. The payments received are not refundable and are generally based on a contractual cost per full-time equivalent employee working on the project. Research and development expenses under the collaborative research agreements approximate or exceed the revenue recognized under such agreements over the term of the respective agreements. Deferred revenue may result if the Company were not to incur the required level of effort during a specific period in comparison to funds received under the respective contracts.
Milestones are recognized pursuant to collaborative agreements upon the achievement of these specified at-risk milestones and upon acknowledgement by the collaborator.
Royalties will be recognized as earned in accordance with the contract terms when the third-party results are reliably measurable and collectibility is reasonably assured.
5. Equity financing
During January 2002, the Company issued 7,000,000 shares of common stock in a registered direct offering to certain institutional investors at a price of $4.50 per share under the Company’s shelf registration statement. The Company received net proceeds of approximately $29.4 million after deducting commissions and offering costs. During February 2002, the Company issued 465,117 shares of common stock in a registered direct offering to a certain institutional investor at a price of $4.30 per share under the Company’s shelf registration statement. The Company received net proceeds of approximately $1.8 million after deducting commissions and offering costs.
6
6. Equipment financing
In January 2002, the Company entered into an additional equipment lease line agreement for an aggregate total of $2.0 million. The Company also issued a warrant to purchase 23,810 shares of common stock at an exercise price of $4.20 per share in conjunction with the agreement. The fair market value of this warrant, as determined by the Black-Scholes valuation model, was approximately $66,000. This amount has been capitalized in other long-term assets and will be amortized into expense over the payment periods of the obligation. As of September 30, 2002, the Company had utilized all of this lease line. The lease period for this facility is three years with the interest rate on each lease line fixed at the time of draw down. As of September 30, 2002, the average interest rate on outstanding obligations was 11.5%. Obligations under this lease line are secured by assets financed under the leases.
7. Daiichi collaboration
In August 2002, the Company entered into a three-year collaborative research agreement with Daiichi Pharmaceuticals (“Daiichi”) to discover and develop drug candidates related to a specific protein degradation target. Under the terms of the agreement, the initial stages of the collaboration will focus on the development of the assay for this specific target and the initiation of high-throughput screening to identify small molecules with therapeutic oncology applications. Upon signing of the agreement, Daiichi was obligated to pay a one-time, non-refundable, non-creditable up-front fee. Under the terms of the agreement, Daiichi will provide support for research for three years, as well as payment for various milestones and royalties if certain conditions are met.
8. Tenant improvements and equipment financing
In July 2002, the Company entered into a tenant improvement and equipment lease line agreement for an aggregate total of $15.0 million. The Company also issued a warrant to purchase 138,889 shares of common stock at an exercise price of $2.70 per share in conjunction with the agreement. The fair market value of this warrant, as determined by the Black-Scholes valuation model, was approximately $251,000. This amount has been capitalized in other long-term assets and will be amortized into expense over the payment periods of the obligation. Due to the master lease agreement amendment signed in October 2002 (see Note 9) the Company is reconsidering the need for the $15.0 million level of financing. Therefore, the Company is currently in negotiations with the lender to amend the agreement to substantially lower the aggregate amount of the line, remove the tenant improvement availability in the line, and remove certain liens that were required under the original agreement. As of September 30, 2002, the Company did not have access to any amount under the original tenant improvement and equipment lease line.
9. Subsequent event
On October 19, 2002, the Company amended its original master lease agreement for future research and office facilities, consisting of approximately 147,000 square feet in South San Francisco, California. The terms of the amendment provide for a delay of the rent commencement date until February 1, 2003, an increase in the tenant improvement allowance to cover the remaining construction obligations on the new facility, and an increase in future rental commitments to compensate for the delay of the rent commencement and the increase in the tenant improvement allowance. The Company also issued a warrant to purchase 500,000 shares of common stock at an exercise price of $1.97 per share in conjunction with the agreement. The fair market value of this warrant, as determined by the Black-Scholes valuation model, will be capitalized in other long-term assets and amortized into expense over the life of the amended lease.
7
Independent Accountants' Review Report
The Board of Directors
Rigel Pharmaceuticals, Inc.
We have reviewed the accompanying condensed balance sheet of Rigel Pharmaceuticals, Inc. as of September 30, 2002, and the related condensed statements of operations for the three-month and nine-month periods ended September 30, 2002 and 2001, and the condensed statements of cash flows for the nine-month periods ended September 30, 2002 and 2001. These financial statements are the responsibility of the Company's management.
We conducted our review in accordance with standards established by the American Institute of Certified Public Accountants. A review of interim financial information consists principally of applying analytical procedures to financial data, and making inquiries of persons responsible for financial and accounting matters. It is substantially less in scope than an audit conducted in accordance with auditing standards generally accepted in the United States, which will be performed for the full year with the objective of expressing an opinion regarding the financial statements taken as a whole. Accordingly, we do not express such an opinion.
Based on our review, we are not aware of any material modifications that should be made to the accompanying condensed financial statements referred to above for them to be in conformity with accounting principles generally accepted in the United States.
We have previously audited, in accordance with auditing standards generally accepted in the United States, the balance sheet of Rigel Pharmaceuticals, Inc. as of December 31, 2001, and the related statements of operations, stockholders' equity, and cash flows for the year then ended (not presented herein) and in our report dated January 25, 2002, (except for note 9, as to which the date is February 20, 2002), we expressed an unqualified opinion on those financial statements. In our opinion, the information set forth in the accompanying condensed balance sheet as of December 31, 2001, is fairly stated, in all material respects, in relation to the balance sheet from which it has been derived.
/s/ ERNST & YOUNG LLP
Palo Alto, California
October 15, 2002
8
Item 2. Management’s Discussion and Analysis of Financial Condition and Results of Operations
This discussion and analysis should be read in conjunction with our financial statements and accompanying notes included in this report and the 2001 audited financial statements and accompanying notes included in our 2001 Annual Report on Form 10-K. Operating results for the three and nine months ended September 30, 2002 are not necessarily indicative of results that may occur in future periods.
Except for the historical information contained herein, the following discussion contains forward-looking statements that are based upon current expectations. Forward-looking statements involve risks and uncertainties. When used herein, the words “believe,” “anticipate,” “expect,” “estimate” and similar expressions are intended to identify such forward-looking statements. There can be no assurance that these statements will prove to be correct. Our actual results and the timing of events could differ significantly from those discussed here. Factors that could cause or contribute to such differences include, but are not limited to, those discussed in “Risk Factors,” as well as those discussed elsewhere in this report and in our 2001 Annual Report on Form 10-K as filed with the SEC. Rigel undertakes no obligation to update any of the forward-looking statements contained herein to reflect any future events or developments.
Overview
Our mission is to become a source of novel, small-molecule drugs to meet large, unmet medical needs. Our business model is to develop a portfolio of drug candidates and to take these through phase II clinical trials, after which we intend to seek commercialization partners for completion of clinical evaluation, regulatory approval and marketing. We have incurred net losses since inception and expect to incur substantial and increasing losses for the next several years as we continue to move drug candidates into and through preclinical and clinical stages of drug development and expand our research and development activities. To date, we have funded our operations primarily through the sale of equity securities, non-equity payments from collaborative partners and capital asset lease financings.
We received our first funding from our collaborative partners in December 1998. As of September 30, 2002, including both research funding and equity investments, we have received cash proceeds totaling an aggregate of $74.8 million from our collaborative partners, including $12.7 million in the nine months ended September 30, 2002. As of September 30, 2002, our accumulated deficit was approximately $106.7 million.
We expect our sources of revenue for the next several years to consist primarily of payments under our current and future corporate collaborations. Under these arrangements, sources of revenue may include up-front payments, funded research, milestone payments and royalties. The process of carrying out our research programs for our collaborative partners and the development of our own non-partnered products to the later stages of development will require significant additional research and development expenditures, including preclinical testing and clinical trials. These activities, together with our general and administrative expenses, are expected to result in substantial operating losses for the foreseeable future. We will not receive product revenue unless we or our collaborative partners complete clinical trials, obtain regulatory approval and successfully commercialize one or more of our products.
To date, we have entered into collaborations with four major pharmaceutical companies: Johnson & Johnson, Pfizer, Novartis and Daiichi. Johnson & Johnson, Pfizer and Novartis have contributed nearly all of our revenues over the last three years.
In July 2001, we expanded our collaboration with Novartis with the initiation of our angiogenesis program, the fourth and final program in our Novartis collaboration. Pursuant to the expanded Novartis collaboration, we received a $4.0 million up-front payment from Novartis, which will be recognized as revenue ratably over the life of the contract. In addition, the expanded collaboration provides that the angiogenesis research program will be carried out at Rigel, and provides for research reimbursement over the next three years and includes potential future milestones and royalty payments to Rigel. In conjunction with the original collaboration, Novartis paid $4.0 million for 2,000,000 shares of our series D preferred stock that converted to 2,000,000 shares of common stock upon the completion of our initial public offering. The original collaboration also allowed for an additional equity investment by Novartis of up to $10.0 million that was callable by us until our initial public offering. We exercised this right and sold to Novartis 1,428,571 shares of common stock at $7.00 per share concurrent with the closing of the our initial public offering. As of April 16, 2002, Novartis still held all 3,428,571 of these shares.
In May 2002, Novartis elected to conclude the research phases of our two initial joint projects in the autoimmunity and transplant rejection areas, after 42 months each, effective in November 2002 and February 2003, respectively. Pursuant to the collaboration agreement, Novartis had the option to end the research phase on these programs after either 24 months or 42 months.
In December 2001, Johnson & Johnson elected to extend the research phase of our collaboration for an additional two years, resulting in additional research reimbursement through the end of 2003 of approximately $5.0 million.
In February 2002, the research phase of our collaboration with Pfizer concluded with Pfizer accepting a total of seven validated targets. Under our collaboration with Pfizer, these validated targets will continue through the drug discovery and development process at Pfizer.
In August 2002, we initiated a three-year collaborative research agreement with Daiichi Pharmaceuticals to discover and develop drug candidates related to a specific protein degradation target. Under the terms of the agreement, the initial stages of the collaboration
9
will focus on the development of the assay for this specific target and the initiation of high-throughput screening to identify small molecules with therapeutic oncology applications. Upon signing of the agreement, Daiichi was obligated to pay a one-time, non-refundable, non-creditable up-front fee. Under the terms of the agreement, Daiichi will provide support for research for three years, as well as payment for various milestones and royalties if certain conditions are met.
A summary of these partnerships is as follows:
|
Partner |
|
Research Program |
|
Commencement Date |
|
Research Phase End Date |
|
Johnson & Johnson |
|
Tumor Growth—Cell Cycle Inhibition |
|
December 4, 1998 |
|
December 2003 |
|
Pfizer |
|
Asthma/Allergy—IgE Production in B Cells |
|
January 31, 1999 |
|
February 2002 |
|
Novartis |
|
Transplant Rejection—T Cell Activation |
|
May 26, 1999 |
|
November 2002 |
|
Novartis |
|
Autoimmunity Disease—B Cell Activation |
|
August 1, 1999 |
|
February 2003 |
|
Novartis |
|
Chronic Bronchitis (conducted at Novartis) |
|
January 1, 2000 |
|
Ongoing |
|
Novartis |
|
Tumor Growth—Inhibition of Tumor Angiogenesis |
|
July 6, 2001 |
|
July 2004 |
|
Daiichi |
|
Tumor Growth—Protein Degradation Oncology Target |
|
August 1, 2002 |
|
August 2005 |
Under the terms of these collaborations, our partners have agreed to provide up to approximately $13.8 million in future research funding over the next three years, $2.0 million of which is subject to possible cancellation. The cancellation amount relates to the Novartis Tumor Growth research program which allows for the research phase of the program to be terminated by Novartis after 24 months. In addition, we may receive additional payments upon the achievement of specific research and development milestones and royalties upon commercialization of any products.
In order to maintain and increase proceeds from collaborations, we are exploring new opportunities with existing and new potential collaborators. Our earliest partnerships focused on the early stages of drug discovery, specifically on target discovery and validation, while our collaboration with Johnson & Johnson has been expanded to also include both chemistry and compound high-throughput screening, and our recent collaboration with Daiichi focuses on drug discovery and development. We currently anticipate that we will self-fund, at an increased rate of spending, our own research programs to later stages of development prior to partnering with collaborative partners. Therefore, it is expected that future collaborative partnerships may have an expanded focus and could include high-throughput screening, combinatorial and medicinal chemistry, pre-clinical evaluations and/or clinical development. For some programs, we may also seek to enter into collaborations for the development of compounds that we have discovered. The timing, the amount of funds received and the scope of any new collaboration are uncertain, and any compound collaboration will depend on the successful progress of clinical trials. New, expanded or larger collaborations will also be necessary to offset any decrease in proceeds as collaborations come to the end of their terms. Our Novartis programs are multiple-year agreements with the research phases terminating in 2002, 2003 and 2004, while the Johnson & Johnson collaboration concludes its research phase at the end of 2003. As each collaboration reaches the conclusion of its research phase, the parties may evaluate the status of the collaboration and, if appropriate, seek to extend the research phase of the collaboration agreement or negotiate alternative terms.
In June 2002, we resolved a dispute with Inoxell A/S (formed as a spinout from Pharmexa – formally M&E Biotech) by entering into a global patent settlement concerning certain drug target identification technologies, which includes both cross-licensing and joint ownership to certain patents and allows for worldwide freedom of operation for both companies.
Critical Accounting Policies and the Use of Estimates
Our discussion and analysis of our financial condition and results of operations is based upon our financial statements, which have been prepared in accordance with accounting principles generally accepted in the United States. The preparation of these financial statements requires us to make estimates and judgments that affect the reported amounts of assets, liabilities, revenues and expenses, and related disclosure of contingent assets and liabilities. On an on-going basis, we evaluate our estimates, including those related to terms of the research collaborations, investments, stock compensation, impairment issues, the estimated useful life of assets, income taxes, financing operations and contingencies. We base our estimates on historical experience and on various other assumptions that we believe to be reasonable under the circumstances, the results of which form the basis for making judgments about the carrying values of assets and liabilities that are not readily apparent from other sources. Actual results may differ from these estimates under different assumptions or conditions. We believe the following critical accounting policies affect our more significant judgments and estimates used in the preparation of our financial statements.
Revenue Recognition
Non-refundable, up-front payments received in connection with research and development collaboration agreements, including technology access fees, are deferred and recognized on a straight-line basis over the relevant periods of continuing involvement, generally the research term.
Revenues related to collaborative research with our corporate collaborators are recognized as research services are performed over the related funding periods for each contract. We are required to perform research and development activities as specified in each respective agreement. The payments received are not refundable and are generally based on a contractual cost per full-time equivalent employee working on the project. Research and development expenses under the collaborative research agreements approximate or exceed the revenue recognized under such agreements over the term of the respective agreements. Deferred revenue may result if we were not to incur the required level of effort during a specific period in comparison to funds received under the respective contracts.
10
Revenues resulting from the achievement of milestones are recognized pursuant to collaborative agreements upon the accomplishment of these specified at-risk milestones.
Royalties will be recognized as earned in accordance with the contract terms when the third-party results are reliably measurable and collectibility is reasonably assured.
Stock-based Compensation
We recorded no deferred stock compensation with respect to stock options granted to employees in the first nine months of 2002 and approximately $0.3 million for the nine months ended September 30, 2001, representing the difference between the deemed fair value of our common stock for financial reporting purposes on the date these options were granted and the exercise price. These amounts have been reflected as components of stockholders’ equity, and the deferred expense is being amortized to operations over the vesting period of the options, generally four to five years, using the graded vesting method. We amortized deferred stock compensation of $0.8 million and $2.1 million for the nine months ended September 30, 2002 and 2001, respectively. At September 30, 2002, we had a total of $1.1 million remaining to be amortized over the remaining vesting periods of the stock options.
In addition to the amortization of the deferred stock compensation, we also record charges associated with stock options granted to consultants in accordance with accounting principles generally accepted in the United States that involve the periodic revaluation of outstanding unvested consultant options based upon the current market value of our common stock and other assumptions, including the expected future volatility of our stock price. We recognized $113,000 and $496,000 in stock-based compensation recovery for revaluation of consultant options for the nine months ended September 30, 2002 and 2001, respectively. Even though the number of unvested outstanding options issued to consultants continues to decline, we expect to see continued fluctuations in the future as a portion of these options are revalued based on the current market price of our common stock through the application of the graded vesting method.
Three Months Ended September 30, 2002 and 2001
Revenues. Contract revenues from collaborations were approximately $3.7 million and $4.2 million for the three months ended September 30, 2002 and 2001, respectively. Revenues in both three-month periods primarily consisted of research support and amortization of upfront fees from the continuation of our collaborations with Novartis and Johnson & Johnson. The decrease in 2002 revenues of $0.5 million was primarily due to the conclusion of the research phase of the Pfizer collaboration in February 2002, offset by the initiation of the Daiichi collaboration in August 2002. We expect contract revenues from collaborations to be a significant component of our total revenues for the foreseeable future.
Research and Development. Research and development expenses were approximately $11.6 million and $8.6 million for the three months ended September 30, 2002 and 2001, respectively. The increase of $3.0 million reflects the costs associated with the commencement of clinical trials, the addition of both drug development and research headcount, and increased preclinical activities. In September 2002, we began the Phase I safety trial of our lead compound, labeled R112, in the United Kingdom and expect to file an IND application for this compound with the FDA, before the end of the year for the clinical indication of allergic rhinitis. We expect research and development expenses to increase in future periods, particularly as we continue to move our solely-owned product candidates through pre-clinical activities and into clinical trials.
The scope and magnitude of future research and development expenses are difficult to predict at this time given the number of studies that will need to be conducted for any of our potential products. In general, biopharmaceutical development involves a series of steps—beginning with identification of a potential target and including, among others, proof of concept in animals and Phase I, II and III clinical studies in humans—each of which is typically more expensive than the previous step. Success in development, therefore, results in increasing expenditures. Our research and development expenditures currently include costs for scientific personnel, supplies, equipment, consultants, patent filings, sponsored research and allocated facility costs. Future research and development expenses are also expected to include costs related to clinical trials.
Because of the number of research projects we have ongoing at any one time, and the ability to utilize resources across several projects, the majority of our research and development costs are not directly tied to any individual project and are allocated among multiple projects. The management of our projects is driven primarily by scientific data, and to a lesser extent, by these cost allocations, which are based primarily on human resource time incurred on each project. As a result, the costs allocated to a project do not necessarily reflect the actual costs of the project. Accordingly, we do not maintain actual cost incurred information for our projects on a project-by-project basis.
General and Administrative Expenses. General and administrative expenses were approximately flat at $2.1 million and $2.0 million for the three months ended September 30, 2002 and 2001, respectively. We expect that general and administrative expenses will increase in the future to support the growth of our research and development efforts as our products continue to move into clinical trials.
Net Interest Expense/Income. Net interest expense was approximately $41,000 for the three months ended September 30, 2002, compared to net interest income of $197,000 for the three months ended September 30, 2001. Interest income results from our interest-bearing
11
balances, whereas interest expense is the result of our capital lease obligations associated with fixed asset purchases. A reduction in interest rates on our owned securities is primarily responsible for the net interest expense in the three months ended September 30, 2002, as compared to the net interest income in the same period in 2001.
Nine Months Ended September 30, 2002 and 2001
Revenues. Contract revenues from collaborations were approximately $12.1 million and $10.5 million for the nine months ended September 30, 2002 and 2001, respectively. Revenues in both nine-month periods primarily consisted of research support and amortization of up-front fees from the continuation of our collaborations with Novartis, Johnson & Johnson and Pfizer. In the nine months ended September 30, 2002, revenues included a milestone payment for a target accepted in accordance with our transplant rejection research program with Novartis and targets accepted in accordance with our Pfizer collaboration. The increase in 2002 revenues of $1.6 million was primarily due to the commencement of the angiogenesis program with Novartis in July 2001 and milestones achieved in the Novartis and Pfizer collaborations. We expect contract revenues from collaborations to be a significant component of our total revenues for the foreseeable future.
Research and Development. Research and development expenses were approximately $33.8 million and $23.4 million for the nine months ended September 30, 2002 and 2001, respectively. The increase of $10.4 million reflects primarily the continued expansion of our drug development infrastructure, the addition of both drug development and research headcount, increased outside contract efforts, increased preclinical activities, the commencement of clinical trials and costs associated with our intellectual property. In September 2002, we began the Phase I safety trial of our lead compound, labeled R112, in the United Kingdom and expect to file an IND application for this compound with the FDA before the end of the year for the clinical indication of allergic rhinitis. We expect research and development expenses to increase in future periods, particularly as we continue to move our solely-owned product candidates through pre-clinical activities and into clinical trials.
The scope and magnitude of future research and development expenses are difficult to predict at this time given the number of studies that will need to be conducted for any of our potential products. In general, biopharmaceutical-development involves a series of steps—beginning with identification of a potential target and including, among others, proof of concept in animals and Phase I, II and III clinical studies in humans—each of which is typically more expensive than the previous step. Success in development, therefore results in increasing expenditures. Our research and development expenditures currently include costs for scientific personnel, supplies, equipment, consultants, patent filings, sponsored research and allocated facility costs. Future research and development expenses are also expected to include costs related to clinical trials.
Because of the number of research projects we have ongoing at any one time, and the ability to utilize resources across several projects, the majority of our research and development costs are not directly tied to any individual project and are allocated among multiple projects. The management of our projects is driven primarily by scientific data, and to a lesser extent, by these cost allocations, which are based primarily on human resource time incurred on each project. As a result, the costs allocated to a project do not necessarily reflect the actual costs of the project. Accordingly, we do not maintain actual cost incurred information for our projects on a project-by-project basis.
General and Administrative Expenses. General and administrative expenses were approximately $7.3 million and $5.9 million for the nine months ended September 30, 2002 and 2001, respectively. The increase was primarily attributable to higher employee costs and greater infrastructure costs to support our growing research and development activities. We expect that general and administrative expenses will increase in the future to support the growth of our research and development efforts as our products continue to move into clinical trials.
Net Interest Income. Net interest income was approximately $68,000 and $1,062,000 for the nine months ended September 30, 2002 and 2001, respectively. Interest income results from our interest-bearing balances, whereas interest expense is the result of our capital lease obligations associated with fixed asset purchases. The decrease in net interest income in 2002 was due to the reduction in interest rates on our owned securities.
Liquidity and Capital Resources
We have financed our operations from inception primarily through sales of equity securities, contract payments payable to us under our collaboration agreements and equipment financing arrangements. As of September 30, 2002, we had received $126.1 million in gross proceeds from the sale of equity securities, including $20.0 million from collaborators, and had received $54.8 million in research funding from collaborators. In addition, as of September 30, 2002, we had financed, through leases and loans, the purchase of equipment and leasehold improvements totaling approximately $17.2 million.
As of September 30, 2002, we had approximately $36.5 million in cash, cash equivalents and available-for-sale securities, as compared to $33.4 million as of December 31, 2001, an increase of $3.1 million. The increase was attributable to proceeds of $31.2 million, net of commissions and offering costs, from the sale of 7,465,117 shares of our common stock to certain institutional investors in two offerings in January and February 2002 under our shelf registration statement, offset by approximately $19.9 million in net cash used in operating activities. We also invested $7.9 million in capital equipment and tenant improvements and had debt service payments of $2.7 million in conjunction with our equipment financing arrangements. These payments were offset by $2.0 million of proceeds from lease financings.
As of September 30, 2002, we had $6.7 million in capital lease obligations associated with our financed purchase of equipment and
12
leasehold improvements. All existing equipment financing agreements as of September 30, 2002 are secured by the equipment financed, bear interest rates in a range of 7% to 15% and are due in monthly installments through 2005. In addition, three of these agreements have balloon payments at the end of each loan term, while the fourth agreement allows us to purchase the assets financed at the fair market value or 20% of the original acquisition cost at the end of the financing term. In July 2002, we entered into a tenant improvement and equipment lease line agreement for an aggregate total of $15.0 million. Due to the master lease agreement amendment signed in October 2002 we are reconsidering the need for the $15.0 million level of financing. Therefore, we are currently in negotiations with the lender to amend the agreement to substantially lower the aggregate amount of the line, remove the tenant improvement availability in the line, and remove certain liens that were required under the original agreement. As of September 30, 2002, we did not have access to any amount under the original tenant improvement and equipment lease line.
In May 2001, we entered into a 15-year non-cancelable lease for future research and office facilities, consisting of approximately 147,000 square feet in South San Francisco, California. On October 19, 2002, we amended the lease. The terms of the amendment allow for a delay of the rent commencement date until February 1, 2003, an increase in the tenant improvement allowance to cover the remaining construction obligations on the new facility, and an increase in future rental commitments to compensate for the delay of the rent commencement and the increase in the tenant improvement allowance.
Under the terms of this amended lease, we are expected to occupy these new facilities in early 2003 and concurrently terminate the lease of our current facilities at 240 East Grand Avenue in South San Francisco. The future research and office facilities are currently under construction as a build-to-suit facility. We have incurred approximately $6.6 million in pre-construction and construction costs associated with our new facility through September 30, 2002. We do not expect to incur any further construction costs due to the increased tenant improvement allowance in the amended lease. These costs are currently being capitalized on our balance sheet as construction-in-progress, a component of property and equipment. These leasehold improvements will be amortized ratably over the term of the lease, which is 15 years, upon occupation of the buildings.
The following are our contractual commitments (by fiscal year) as of October 20, 2002 associated with debt obligations, lease obligations, contracted research obligations and tenant improvement obligations:
|
|
|
Total |
|
2003 |
|
2004 - 2005 |
|
2006 - 2007 |
|
2008 - 2018 |
|
|||||
|
|
|
(in thousands) |
|
|||||||||||||
|
Capital leases |
|
$ |
6,641 |
|
$ |
3,977 |
|
$ |
2,664 |
|
$ |
— |
|
$ |
— |
|
|
Facilities leases |
|
198,128 |
|
7,169 |
|
21,438 |
|
26,546 |
|
142,975 |
|
|||||
|
Contracted research |
|
500 |
|
500 |
|
— |
|
— |
|
— |
|
|||||
|
Total |
|
$ |
205,269 |
|
$ |
11,646 |
|
$ |
24,102 |
|
$ |
26,546 |
|
$ |
142,975 |
|
We believe that our existing capital resources, together with the proceeds from our current collaborations as well as proceeds from future anticipated collaborations, will be sufficient to support our current operating plan for at least the next 12 months. We will require additional financing in the future to fund our operations. Our future funding requirements will depend on many factors, including, but not limited to:
• our ability to maintain our existing collaboration partnerships;
• our ability to establish, and the scope of, new collaborations;
• the progress and number of research programs carried out at Rigel;
• the progress of the research and development efforts of our collaborators;
• any changes in the breadth of our research and development programs;
• our ability to meet the milestones identified in our collaborative agreements that trigger payments;
• our ability to maintain and establish new corporate relationships and research collaborations;
• our ability to acquire or license other technologies or compounds, if any;
• the progress and success of preclinical studies and clinical trials of our drug candidates conducted by us or our collaborative partners or licensees;
• our ability to manage our growth;
13
• competing technological and market developments;
• the costs and timing of obtaining, enforcing and defending our patent and intellectual rights;
• the costs and timing of regulatory approvals; and
• expenses associated with unforeseen litigation.
In addition, we are constantly reviewing potential opportunities to expand our technologies or add to our portfolio of drug candidates. In the future, we may need further capital in order to acquire or invest in technologies, products or businesses. For the next several years, we do not expect our operations to generate the amount of cash required by our future cash needs. In December 2001, we filed a registration statement on Form S-3 to offer and sell equity and debt securities in one or more offerings up to a total dollar amount of $50 million. Currently, approximately $16.5 million remains available on the Form S-3, and we have no current commitments to offer and sell any securities that may be offered and sold pursuant to such registration statement. We expect to finance future cash needs through strategic collaborations, strategic financings, debt financing, and the sale of equity securities. We cannot assure you that additional financing or collaboration and licensing arrangements will be available when needed or that, if available, such financing will be obtained on terms favorable to us or our stockholders. Insufficient funds may require us to delay, scale back or eliminate some or all of our research or development programs, to lose rights under existing licenses or to relinquish greater or all rights to product candidates at an earlier stage of development or on less favorable terms than we would otherwise choose or may adversely affect our ability to operate as a going concern. If additional funds are obtained by issuing equity securities, substantial dilution to existing stockholders may result.
Risk Factors
An investment in our securities is risky. Prior to making a decision about investing in our securities you should carefully consider the following risks, as well as the other information contained in this quarterly report filed on Form 10-Q. If any of the following risks actually occurs, our business could be harmed. In that case, the trading price of our common stock could decline, and you might lose all or part of your investment. The risks and uncertainties described below are not the only ones facing us. Additional risks and uncertainties not presently known to us, or that we currently see as immaterial, may also harm our business. If any of these additional risks or uncertainties occurs, the trading price of our common stock could decline, and you might lose all or part of your investment.
Our success as a company is uncertain due to our limited operating history, our history of operating losses and the uncertainty of future profitability.
Due in large part to the significant research and development expenditures required to identify and validate new drug candidates and advance our programs into clinical testing, we have not been profitable and have generated operating losses since we were incorporated in June 1996. Currently, our revenues are generated solely from research payments from our collaboration agreements and licenses and are insufficient to generate profitable operations. As of September 30, 2002, we had an accumulated deficit of approximately $106.7 million. We expect to incur losses for at least the next several years and expect that these losses will actually increase as we expand our research and development activities, incur significant clinical and testing costs and expand our facilities. Moreover, our losses are expected to continue even if our current research projects are able to successfully identify potential drug targets. If the time required to generate revenues and achieve profitability is longer than anticipated or if we are unable to obtain necessary capital, we may not be able to fund and continue our operations.
Because most of our expected future revenues are contingent upon collaborative and license agreements, we might not meet our strategic objectives.
Our ability to generate revenues in the near term depends on our ability to enter into additional collaborative agreements with third parties and to maintain the agreements we currently have in place. Our ability to enter into new collaborations and the revenue, if any, that may be recognized under these collaborations is highly uncertain. If we are unable to enter into new collaborations, our business prospects could be harmed, which could have an immediate adverse effect on the trading price of our stock.
To date, most of our revenues have been related to the research phase of each of our collaborative agreements. Such revenues are for specified periods, and the impact of such revenues on our results of operations is partially offset by corresponding research costs. Following the completion of the research phase of each collaborative agreement, additional revenue may come only from milestone payments and royalties, which may not be paid, if at all, until some time well into the future. The risk is heightened due to the fact that unsuccessful research efforts may preclude us from receiving any milestone payments under these agreements. Our receipt of revenue from collaborative arrangements is also significantly affected by the timing of efforts expended by us and our collaborators and the timing of lead compound identification. In late 2001, we recorded the first revenue from achievement of milestones in both the Pfizer and Johnson & Johnson collaborations. Under many agreements, milestone payments may not be earned until the collaborator has advanced products into clinical testing, which may never occur or may not occur until some time well into the future. We may not recognize revenue under our collaborations when and in accordance with our expectations or the expectations of industry analysts, which could harm our business and
14
have an immediate adverse effect on the trading price of our stock.
Our business plan forsees that we will need to generate meaningful revenue from royalties and licensing agreements. To date, we have not yet received any revenue from royalties for the sale of commercial drugs, and we do not know when we will receive any such revenue, if at all. Likewise, we have not licensed any lead compounds or drug development candidates to third parties, and we do not know whether any such license will be entered into on acceptable terms in the future, if at all.
We are unable to predict when, or if, we will become profitable, and even if we are able to achieve profitability at any point in time, we do not know if our operations will be able to maintain profitability during any future periods.
There is a high risk that early-stage drug discovery and development might not successfully generate good drug candidates.
At the present time, the majority of our operations are in the early stages of drug identification and development. To date, we have only identified a few potential drug compounds and only one of those compounds has made it into the clinical testing stage. In our industry, it is statistically unlikely that the few compounds that we have identified as potential drug candidates will actually lead to successful drug development efforts, and we do not expect any drugs resulting from our research to be commercially available for several years, if at all. Our one product in the clinic and our future leads for potential drug compounds will be subject to the risks and failures inherent in the development of pharmaceutical products based on new technologies. These risks include, but are not limited to, the inherent difficulty in selecting the right drug target and avoiding unwanted side effects as well as the unanticipated problems relating to product development, testing, regulatory compliance, manufacturing, marketing and competition and additional costs and expenses that may exceed current estimates.
We might not be able to commercialize our drug candidates successfully if problems arise in the testing and approval process.
Commercialization of our product candidates depends upon successful completion of preclinical studies and clinical trials. Preclinical testing and clinical development are long, expensive and uncertain processes. We do not know whether we, or any of our collaborative partners, will be permitted to undertake clinical trials of potential products beyond the one trial already concluded. It may take us or our collaborative partners several years to complete any such testing, and failure can occur at any stage of testing. Interim results of trials do not necessarily predict final results, and acceptable results in early trials may not be repeated in later trials. A number of companies in the pharmaceutical industry, including biotechnology companies, have suffered significant setbacks in advanced clinical trials, even after achieving promising results in earlier trials. Moreover, when our projects reach clinical trials, we or our collaborative partners may decide to discontinue development of any or all of these projects at any time for commercial, scientific or other reasons. There is also a risk that competitors and third parties may develop similar or superior products or have proprietary rights that preclude us from ultimately marketing our products, as well as the potential risk that our products may not be accepted by the marketplace.
If our current corporate collaborations or license agreements are unsuccessful or if conflicts develop with these relationships, our research and development efforts could be delayed.
Our strategy depends upon the formation and sustainability of multiple collaborative arrangements and license agreements with third parties in the future. We rely on these arrangements for not only financial resources, but also for expertise that we expect to need in the future relating to clinical trials, manufacturing, sales and marketing, and for licenses to technology rights. To date, we have entered into several such arrangements with corporate collaborators; however, we do not know if such third parties will dedicate sufficient resources or if any such development or commercialization efforts by third parties will be successful. Should a collaborative partner fail to develop or commercialize a compound or product to which it has rights from us, we may not receive any future milestone payments and will not receive any royalties associated with such compound or product. In addition, the continuation of some of our partnered drug discovery and development programs may be dependent on the periodic renewal of our corporate collaborations. For example, the funded research phase of our collaboration with Pfizer has been completed and the development portion of our collaboration is ongoing at Pfizer. In addition, in May 2002, Novartis elected to conclude the research phases of our two initial joint projects in the autoimmunity and transplant rejection areas, after 42 months each, effective February 2003. Pursuant to the collaboration agreement, Novartis had the option to end the research phase on these programs after 24 months or 42 months. More generally, our corporate collaboration agreements may terminate before the full term of the collaborations or upon a breach or a change of control. We may not be able to renew these collaborations on acceptable terms, if at all, or negotiate additional corporate collaborations on acceptable terms, if at all.
We are also a party to various license agreements that give us rights to use specified technologies in our research and development processes. The agreements, pursuant to which we have in-licensed technology, permit our licensors to terminate the agreements under certain circumstances. If we are not able to continue to license these and future technologies on commercially reasonable terms, our product development and research may be delayed.
Conflicts might also arise with respect to our various relationships with third parties. If any of our corporate collaborators were to breach or terminate its agreement with us or otherwise fail to conduct the collaborative activities successfully and in a timely manner, the preclinical or clinical development or commercialization of the affected product candidates or research programs could be delayed or terminated. We generally do not control the amount and timing of resources that our corporate collaborators devote to our programs or
15
potential products. We do not know whether current or future collaborative partners, if any, might pursue alternative technologies or develop alternative products either on their own or in collaboration with others, including our competitors, as a means for developing treatments for the diseases targeted by collaborative arrangements with us. Conflicts also might arise with collaborative partners concerning proprietary rights to particular compounds. While our existing collaborative agreements typically provide that we retain milestone payments and royalty rights with respect to drugs developed from certain derivative compounds, any such payments or royalty rights may be at reduced rates, and disputes may arise over the application of derivative payment provisions to such drugs, and we may not be successful in such disputes.
If we fail to enter into new collaborative arrangements in the future, our business and operations would be negatively impacted.
Although we have established several collaborative arrangements and various license agreements, we do not know if we will be able to establish additional arrangements, or whether current or any future collaborative arrangements will ultimately be successful. For example, there have been, and may continue to be, a significant number of recent business combinations among large pharmaceutical companies that have resulted, and may continue to result, in a reduced number of potential future corporate collaborators, which may limit our ability to find partners who will work with us in developing and commercializing our drug targets. If business combinations involving our existing corporate collaborators were to occur, the effect could be to diminish, terminate or cause delays in one or more of our corporate collaborations.
We will need additional capital in the future to sufficiently fund our operations and research.
Our operations require significant additional funding in large part due to our research and development expenses, future preclinical and clinical-testing costs, the expansion of our facilities and the absence of any meaningful revenues over the foreseeable future. The amount of future funds needed will depend largely on the success of our collaborations and our research activities, and we do not know whether additional financing will be available when needed, or that, if available, we will obtain financing on terms favorable to our stockholders or us. We have consumed substantial amounts of capital to date, and operating expenditures are expected to increase over the next several years as we expand our infrastructure and research and development activities.
We believe that our existing capital resources, together with the proceeds from our current collaborations as well as proceeds from future anticipated collaborations, will be sufficient to support our current operating plan for at least the next 12 months. We will require additional financing in the future to fund our operations. Our future funding requirements will depend on many factors, including, but not limited to:
• our ability to maintain our existing collaboration partnerships;
• our ability to establish and the scope of new collaborations;
• the progress and number of research programs carried out at Rigel;
• the progress of the research and development efforts of our collaborators;
• any changes in the breadth of our research and development programs;
• our ability to meet the milestones identified in our collaborative agreements that trigger payments;
• our ability to maintain and establish new corporate relationships and research collaborations;
• our ability to acquire or license other technologies or compounds, if any;
• the progress and success of preclinical studies and clinical trials of our drug candidates conducted by us or our collaborative partners or licensees;
• our ability to manage our growth;
• competing technological and market developments;
• the costs and timing of obtaining, enforcing and defending our patent and intellectual rights;
• the costs and timing of regulatory approvals; and
• expenses associated with unforeseen litigation.
To the extent we raise additional capital by issuing equity securities, our stockholders may experience substantial dilution. To the extent that we raise additional funds through collaboration and licensing arrangements, we may be required to relinquish some rights to our technologies or product candidates, or grant licenses on terms that are not favorable to us. If adequate funds are not available, we will not be able to continue developing our products.
Our success is dependent on intellectual property rights held by us and third parties, and our interest in such rights is complex and uncertain.
Our success will depend to a large part on our own, our licensees’ and our licensors’ ability to obtain and defend patents for each party’s respective technologies and the compounds and other products, if any, resulting from the application of such technologies. Twelve U.S. patents have been issued to us as of October 15, 2002, and we have numerous applications in the U.S. and abroad awaiting approval.
16
In the future, our patent position might be highly uncertain and involve complex legal and factual questions. No consistent policy regarding the breadth of claims allowed in biotechnology patents has emerged to date. Accordingly, we cannot predict the breadth of claims allowed in our or other companies’ patents.
The degree of future protection for our proprietary rights is uncertain, and we cannot ensure that:
• we were the first to make the inventions covered by each of our pending patent applications;
• we were the first to file patent applications for these inventions;
• others will not independently develop similar or alternative technologies or duplicate any of our technologies;
• any of our pending patent applications will result in issued patents;
• any patents issued to us or our collaborators will provide a basis for commercially viable products or will provide us with any competitive advantages or will not be challenged by third parties;
• we will develop additional proprietary technologies that are patentable; or
• the patents of others will not have a negative effect on our ability to do business.
We rely on trade secrets to protect technology where we believe patent protection is not appropriate or obtainable. However, trade secrets are difficult to protect. While we require employees, collaborators and consultants to enter into confidentiality agreements, we may not be able to adequately protect our trade secrets or other proprietary information in the event of any unauthorized use or disclosure or the lawful development by others of such information.
We are a party to certain in-license agreements that are important to our business, and we generally do not control the prosecution of in-licensed technology. Accordingly, we are unable to exercise the same degree of control over this intellectual property as we exercise over our internally-developed technology. Moreover, some of our academic institution licensors, research collaborators and scientific advisors have rights to publish data and information in which we have rights. If we cannot maintain the confidentiality of our technology and other confidential information in connection with our collaborations, then our ability to receive patent protection or protect our proprietary information will be impaired. In addition, some of the technology we have licensed relies on patented inventions developed using U.S. government resources. The U.S. government retains certain rights, as defined by law, in such patents, and may choose to exercise such rights.
If a dispute arises regarding the infringement or misappropriation of the proprietary rights of others, such dispute could be costly and result in delays in our research and development activities.
Our success will also depend, in part, on our ability to operate without infringing or misappropriating the proprietary rights of others. There are many issued patents and patent applications filed by third parties relating to products or processes that are similar or identical to ours or our licensors, and others may be filed in the future. There can be no assurance that our activities, or those of our licensors, will not infringe patents owned by others. For example, in June 2002, we resolved a dispute with Inoxell A/S (formed as a spinout from Pharmexa – formally M&E Biotech) by entering into a global patent settlement concerning certain drug target identification technologies, which includes both cross-licensing and joint ownership to certain patents and allows for worldwide freedom of operation for both companies. We believe that there may be significant litigation in the industry regarding patent and other intellectual property rights, and we do not know if we or our collaborators would be successful in any such litigation. Any legal action against our collaborators or us claiming damages or seeking to enjoin commercial activities relating to the affected products, our methods or processes could:
• require our collaborators or us to obtain a license to continue to use, manufacture or market the affected products, methods or processes, which may not be available on commercially reasonable terms, if at all;
• prevent us from using the subject matter claimed in the patents held by others;
• subject us to potential liability for damages;
• consume a substantial portion of our managerial and financial resources; and
• result in litigation or administrative proceedings that may be costly, whether we win or lose.
If we are unable to obtain regulatory approval to market products in the United States and foreign jurisdictions, we might not be permitted to commercialize products from our research and development.
Due, in part, to the early stage of our drug candidate research and development process, we cannot predict whether regulatory clearance will be obtained for any product we, or our collaborative partners, hope to develop. Satisfaction of regulatory requirements typically takes many years, is dependent upon the type, complexity and novelty of the product and requires the expenditure of substantial resources. Of particular significance to us are the requirements covering research and development and testing.
Before commencing clinical trials in humans in the United States, we, or our collaborative partners, will need to submit and receive approval from the FDA of an investigational new drug application (IND). If regulatory clearance of a product is granted, this clearance will be limited to those disease states and conditions for which the product is demonstrated through clinical trials to be safe and
17
efficacious. We cannot ensure that any compound developed by us, alone or with others, will prove to be safe and efficacious in clinical trials and will meet all of the applicable regulatory requirements needed to receive marketing clearance.
Outside the United States, our ability, or that of our collaborative partners, to market a product is contingent upon receiving a marketing authorization from the appropriate regulatory authorities. This foreign regulatory approval process typically includes all of the risks associated with FDA clearance described above and may also include additional risks.
We may encounter difficulties in managing our growth, and these difficulties could increase our losses.
We have experienced a period of rapid and substantial growth that has placed, and will continue to place, a strain on our human and capital resources. The number of our employees increased from 31 at December 31, 1997 to 162 at September 30, 2002. Our ability to manage our operations and growth effectively requires us to continue to use funds to improve our operational, financial and management controls, reporting systems and procedures and to attract and retain sufficient numbers of talented employees. If we are unable to manage this growth effectively, our losses will increase.
If our competitors develop technologies that are more effective than ours, our commercial opportunity will be reduced or eliminated.
The biotechnology and pharmaceutical industries are intensely competitive and subject to rapid and significant technological change. Many of the drugs that we are attempting to discover will be competing with existing therapies. In addition, a number of companies are pursuing the development of pharmaceuticals that target the same diseases and conditions that we are targeting. We face competition from pharmaceutical and biotechnology companies both in the United States and abroad.
Our competitors may utilize discovery technologies and techniques or partner with collaborators in order to develop products more rapidly or successfully than we, or our collaborators, are able to do. Many of our competitors, particularly large pharmaceutical companies, have substantially greater financial, technical and human resources than we do. In addition, academic institutions, government agencies and other public and private organizations conducting research may seek patent protection with respect to potentially competitive products or technologies and may establish exclusive collaborative or licensing relationships with our competitors.
We believe that our ability to compete is dependent, in part, upon our ability to create, maintain and license scientifically-advanced technology and upon our and our strategic partners’ ability to develop and commercialize pharmaceutical products based on this technology, as well as our ability to attract and retain qualified personnel, obtain patent protection or otherwise develop proprietary technology or processes and secure sufficient capital resources for the expected substantial time period between technological conception and commercial sales of products based upon our technology. The failure by us or any of our collaborators in any of those areas may prevent the successful commercialization of our potential drug targets.
Our competitors might develop technologies and drugs that are more effective or less costly than any that are being developed by us or that would render our technology and potential drugs obsolete and noncompetitive. In addition, our competitors may succeed in obtaining the approval of the FDA or other regulatory agencies for drug candidates more rapidly. Companies that complete clinical trials, obtain required regulatory agency approvals and commence commercial sale of their drugs before their competitors may achieve a significant competitive advantage, including certain patent and FDA marketing exclusivity rights that would delay or prevent our ability to market certain products. Any drugs resulting from our research and development efforts, or from our joint efforts with our existing or future collaborative partners, might not be able to compete successfully with competitors’ existing or future products or products under development or obtain regulatory approval in the United States or elsewhere.
Our ability to generate revenues will be diminished if our collaborative partners fail to obtain acceptable prices or an adequate level of reimbursement for products from third-party payors.
The drugs we hope to develop may be rejected by the marketplace due to many factors, including cost. Our ability to commercially exploit a drug may be limited due to the continuing efforts of government and third-party payors to contain or reduce the costs of health care through various means. For example, in some foreign markets, pricing and profitability of prescription pharmaceuticals are subject to government control. In the United States, we expect that there will continue to be a number of federal and state proposals to implement similar government control. In addition, increasing emphasis on managed care in the United States will likely continue to put pressure on the pricing of pharmaceutical products. Cost control initiatives could decrease the price that any of our collaborators would receive for any products in the future. Further, cost control initiatives could adversely affect our collaborators’ ability to commercialize our products and our ability to realize royalties from this commercialization.
Our ability to commercialize pharmaceutical products with collaborators may depend, in part, on the extent to which reimbursement for the products will be available from:
• government and health administration authorities;
18
• private health insurers; and
• other third-party payors.
Significant uncertainty exists as to the reimbursement status of newly-approved healthcare products. Third-party payors, including Medicare, are challenging the prices charged for medical products and services. Government and other third-party payors increasingly are attempting to contain healthcare costs by limiting both coverage and the level of reimbursement for new drugs and by refusing, in some cases, to provide coverage for uses of approved products for disease indications for which the FDA has not granted labeling approval. Third-party insurance coverage may not be available to patients for any products we discover and develop, alone or with collaborators. If government and other third-party payors do not provide adequate coverage and reimbursement levels for our products, the market acceptance of these products may be reduced.
If conflicts arise between our collaborators or advisors and us, any of them may act in their self-interest, which may be adverse to your interests.
If conflicts arise between us and our corporate collaborators or scientific advisors, the other party may act in its self-interest and not in the interest of our stockholders. Some of our corporate collaborators are conducting multiple product development efforts within each disease area that is the subject of the collaboration with us. In some of our collaborations, we have agreed not to conduct, independently or with any third party, any research that is competitive with the research conducted under our collaborations. Our collaborators, however, may develop, either alone or with others, products in related fields that are competitive with the products or potential products that are the subject of these collaborations. Competing products, either developed by our collaborators or to which our collaborators have rights, may result in their withdrawal of support for our product candidates.
If product liability lawsuits are successfully brought against us, we may incur substantial liabilities and may be required to limit commercialization of our products.
The testing and marketing of medical products entail an inherent risk of product liability. If we cannot successfully defend ourselves against product liability claims, we may incur substantial liabilities or be required to limit commercialization of our products. We currently do not have product liability insurance, and our inability to obtain sufficient product liability insurance at an acceptable cost to protect against potential product liability claims could prevent or inhibit the commercialization of pharmaceutical products we develop, alone or with corporate collaborators. We, or our corporate collaborators, might not be able to obtain insurance at a reasonable cost, if at all. While under various circumstances we are entitled to be indemnified against losses by our corporate collaborators, indemnification may not be available or adequate should any claim arise.
Our research and development efforts will be seriously jeopardized, if we are unable to attract and retain key employees and relationships.
Being a small company with only 162 employees as of September 30, 2002, our success depends on the continued contributions of our principal management and scientific personnel and on our ability to develop and maintain important relationships with leading academic institutions, scientists and companies in the face of intense competition for such personnel. In particular, our research programs depend on our ability to attract and retain highly skilled chemists, other scientists, and regulatory and clinical personnel. If we lose the services of any of our personnel, our research and development efforts could be seriously and adversely affected. Although we generally have not experienced problems retaining key employees, our employees can terminate their employment with us at any time. We also expect to encounter increasing difficulty in attracting enough qualified personnel as our operations expand and the demand for these professionals increases, and this difficulty could impede significantly the achievement of our research and development objectives.
We depend on various scientific consultants and advisors for the success and continuation of our research efforts.
We work extensively with various scientific consultants and advisors. The potential success of our drug discovery programs depends, in part, on continued collaborations with certain of these consultants and advisors. We, and various members of our management and research staff, rely on certain of these consultants and advisors for expertise in our research, regulatory and clinical efforts. Our scientific advisors are not employees of ours and may have commitments to, or consulting or advisory contracts with, other entities that may limit their availability to us. We do not know if we will be able to maintain such consulting agreements or that such scientific advisors will not enter into consulting arrangements, exclusive or otherwise, with competing pharmaceutical or biotechnology companies, any of which would have a detrimental impact on our research objectives and could have a material adverse effect on our business, financial condition and results of operations.
If we use biological and hazardous materials in a manner that causes injury or violates laws, we may be liable for damages.
Our research and development activities involve the controlled use of potentially harmful biological materials as well as hazardous materials, chemicals and various radioactive compounds. We cannot completely eliminate the risk of accidental contamination or injury from the use, storage, handling or disposal of these materials. In the event of contamination or injury, we could be held liable for
19
damages that result, and such liability could exceed our resources. We are subject to federal, state and local laws and regulations governing the use, storage, handling and disposal of these materials and specified waste products. The cost of compliance with, or any potential violation of, these laws and regulations could be significant.
Our facilities are located near known earthquake fault zones, and the occurrence of an earthquake or other catastrophic disaster could cause damage to our facilities and equipment, which could require us to cease or curtail operations.
Our facilities are located in the San Francisco Bay Area near known earthquake fault zones and are vulnerable to significant damage from earthquakes. We are also vulnerable to damage from other types of disasters, including fires, floods, power loss, communications failures and similar events. If any disaster were to occur, our ability to operate our business at our facilities would be seriously, or potentially completely, impaired, and our research could be lost or destroyed. In addition, the unique nature of our research activities and of much of our equipment could make it difficult for us to recover from a disaster. The insurance we maintain may not be adequate to cover or losses resulting from disasters or other business interruptions.
If our officers, directors and largest stockholders choose to act together, they may be able to significantly affect our management and operations, acting in their best interests and not necessarily those of other stockholders.
Our directors, executive officers and principal stockholders and their affiliates beneficially own approximately 47% of our common stock, based on their beneficial ownership as of April 16, 2002. Accordingly, they collectively will have the ability to significantly affect the election of all of our directors and the outcome of most corporate actions requiring stockholder approval. They may exercise this ability in a manner that advances their best interests and not necessarily those of other stockholders.
Our stock price may be volatile, and your investment in our stock could decline in value.
The market prices for our securities and those of other biotechnology companies have been highly volatile and may continue to be highly volatile in the future. The following factors, in addition to other risk factors described in this section, may have a significant impact on the market price of our common stock:
• announcements of technological innovations or new commercial products by our competitors or us;
• developments concerning proprietary rights, including patents;
• developments concerning our collaborations;
• publicity regarding actual or potential medical results relating to products under development by our competitors or us;
• regulatory developments in the United States and foreign countries;
• litigation;
• economic and other external factors or other disaster or crisis; and
• period-to-period fluctuations in financial results.
Anti-takeover provisions in our charter documents and under Delaware law may make an acquisition of us, which may be beneficial to our stockholders, more difficult.
Provisions of our amended and restated certificate of incorporation and bylaws, as well as provisions of Delaware law, could make it more difficult for a third party to acquire us, even if doing so would benefit our stockholders. These provisions:
• establish that members of the board of directors may be removed only for cause upon the affirmative vote of stockholders owning at least two-thirds of our capital stock;
• authorize the issuance of “blank check” preferred stock that could be issued by our board of directors to increase the number of outstanding shares and thwart a takeover attempt;
• limit who may call a special meeting of stockholders;
• prohibit stockholder action by written consent, thereby requiring all stockholder actions to be taken at a meeting of our stockholders;
• establish advance notice requirements for nominations for election to the board of directors or for proposing matters that can be acted upon at stockholder meetings; and
• provide for a board of directors with staggered terms.
In addition, Section 203 of the Delaware General Corporation Law, which imposes certain restrictions relating to transactions with major stockholders, may discourage, delay or prevent a third party from acquiring us.
20
Item 3. Quantitative and Qualitative Disclosures About Market Risk
The primary objective of our investment activities is to preserve principal while at the same time maximizing the income we receive from our investments without significantly increasing risk. Some of the securities in which we invest may have market risk. This means that a change in prevailing interest rates may cause the fair value amount of the investment to fluctuate. For example, if we hold a security that was issued with a fixed interest rate at the then-prevailing rate and the prevailing interest rate later rises, the market value amount of our investment will decline. To minimize this risk in the future, we intend to maintain our portfolio of cash equivalents and short-term investments in a variety of securities, including commercial paper, money market funds and government and non-government debt securities. In 2001 and the first nine months of 2002, we maintained an investment portfolio primarily in depository accounts and corporate commercial paper. Due to the short-term nature of these investments, we believe we do not have a material exposure to interest rate risk arising from our investments. Therefore, no quantitative tabular disclosure is provided.
We have operated primarily in the United States, and all funding activities with our collaborators to date have been made in U.S. dollars. Accordingly, we have not had any exposure to foreign currency rate fluctuations.
Item 4. Controls and Procedures
Evaluation of Disclosure Controls and Procedures. Rigel’s chief executive officer and chief financial officer have concluded that Rigel’s disclosure controls and procedures (as defined in Securities Exchange Act Rule 13a-14(c)) are sufficiently effective to ensure that the information required to be disclosed by the company in the reports it files under the Exchange Act is gathered, analyzed and disclosed with adequate timeliness, accuracy and completeness, based on an evaluation of such controls and procedures conducted within 90 days prior to the date hereof.
Changes in Internal Controls. There have been no significant changes in Rigel’s internal controls or in other factors that could significantly affect these controls subsequent to the date of the evaluation referred to above, nor were there any significant deficiencies or material weaknesses in Rigel’s internal controls. Accordingly, no corrective actions were required or undertaken.
Limitations on the Effectiveness of Controls. A control system, no matter how well conceived and operated, can provide only reasonable, not absolute, assurance that the objectives of the control are met. Because of the inherent limitations in all control systems, no evaluation of controls can provide absolute assurance that all control issues, if any, within a company have been detected.
21
PART II OTHER INFORMATION
Item 2. Changes in Securities and Use of Proceeds
c) On July 12, 2002, we issued a warrant to purchase 138,889 shares of common stock at an exercise price of $2.70 per share to Comerica Bank – California in connection with the execution of a tenant improvement and equipment lease line agreement. The warrant was issued in a private transaction pursuant to an exemption from registration in reliance upon Section 4(2) of the Securities Act of 1933, as amended.
d) Our Registration Statement on Form S-1 (No. 333-45864), as amended, with respect to our initial public offering was declared effective by the SEC on November 28, 2000. We received net proceeds of approximately $35,560,000 after deducting offering expenses of $3,990,000, including underwriting discounts and commissions of $2,768,000 and other offering expenses of $1,222,000. We intend to continue to use the net proceeds of the offering for research and development, general corporate purposes and working capital and capital lease obligations. Rigel continually assesses the specific uses and allocations for these funds. As of September 30, 2002, approximately $4.9 million of the net proceeds remained available and were primarily invested in short-term marketable securities.
Item 5. Other Information
Except for the historical information contained herein, the following discussion contains forward-looking statements that are based upon current expectations. Forward-looking statements involve risks and uncertainties. When used herein, the words “believe,” “anticipate,” “expect,” “estimate” and similar expressions are intended to identify such forward-looking statements. There can be no assurance that these statements will prove to be correct. Our actual results and the timing of events could differ significantly from those discussed here. Factors that could cause or contribute to such differences include, but are not limited to, those discussed in “Risk Factors,” as well as those discussed elsewhere in this report and in our 2001 Annual Report on Form 10-K as filed with the SEC. Rigel undertakes no obligation to update any of the forward-looking statements contained herein to reflect any future events or developments.
Product Development
The following table summarizes key information in the twelve programs being conducted by Rigel and its partners that focus on specific disease mechanisms:
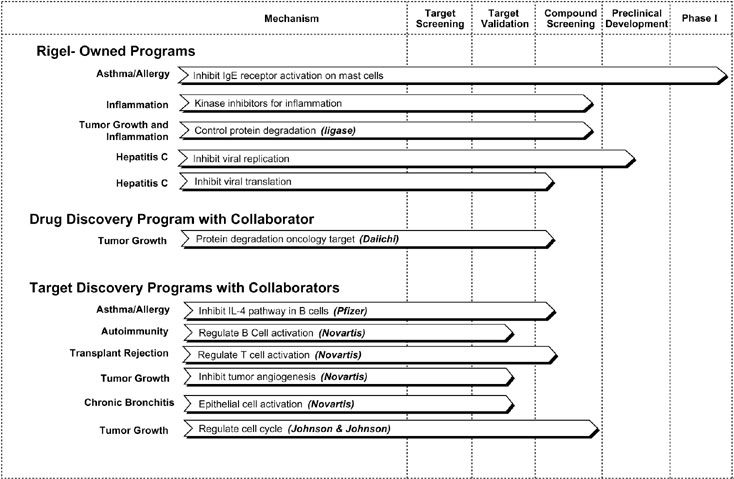
(1) “Target Screening”: Disease-modeled screening in cells using our advanced functional genomics technology.
(2) “Target Validation”: Testing to establish a causal link between an intracellular protein target and a cellular response important in a disease process.
22
(3) “Compound Screening”: The development of biochemical and cell-based assays for the purpose of screening collections of small molecule compounds to identify any compounds that bind to a functionally active site of a validated target and to determine which of those compounds to move into preclinical development.
(4) “Preclinical Development”: Pharmacology and toxicology testing in animal models to gather data necessary to comply with applicable regulatory protocols prior to submission of an Investigational New Drug application to the FDA.
(5) “Phase I”: Clinical testing in humans to determine toxicity.
Immune Disorders
Many diseases and disorders result from defects in the immune system. Over 40 million people in the United States suffered from allergic disorders and over 20 million from asthmatic disorders in 2001. Anti-asthmatic and allergy relief medications exceeded $5 billion in worldwide sales in 2001. In 2001, another 3 million to 5 million patients in the United States were treated for other immune disorders. We currently have programs in immunology focused on asthma/allergy, autoimmunity/transplant rejection, inflammatory diseases, and chronic bronchitis.
Asthma/Allergy
Rigel-Owned Asthma/Allergy Programs
Inhibit IgE receptor activation on mast cells. The goal of this program is to identify compounds that inhibit the secretion of inflammatory factors resulting from IgE binding to its receptor on mast cells. Currently, we have identified several candidate compound series for development. Preliminary studies demonstrate that these compounds inhibit the ability of IgE to activate its receptor on mast cells. There is evidence in animal models and early clinical studies that blocking IgE mediated activation of mast cells can reduce allergic symptoms in multiple species, including humans. We believe that small molecule inhibitors of IgE signaling pathways could play an important role in the treatment of such chronic disorders. We have completed the Phase I safety trial on our lead compound labeled R112 in the United Kingdom and expect to file an investigational new drug, or IND, application for this compound with the United States Food and Drug Administration, or FDA, before the end of the year for the clinical indication of allergic rhinitis.
Asthma/Allergy Program with Pfizer
Inhibit IL-4 production in B Cells. In this program with Pfizer that began in 1999, we have completed work to identify and validate intracellular drug targets that selectively control the production of IgE in B Cells. The program has generated seven validated targets that have been accepted by Pfizer, and several of these targets are now entering the compound screening phase at Pfizer.
Autoimmunity/Transplant Rejection
Autoimmunity disorders and organ transplant rejection are the result of inappropriate activation of the immune system. Most existing therapies also have toxic side effects. A challenge facing all research groups in this field has been the design of selective and specific immune system therapeutics that affect only the pathological activities without negatively affecting the protective activities of the immune system.
Rigel-Owned Kinase Inhibitor for Inflammation
Activation pathways are initiated by the binding of antigen (foreign protein) to specific surface receptors on T cells or B cells. This sets off an intracellular cascade of signals, resulting in changes in gene expression and the production of proteins that drive the immune response or lead to antibody production and secretion in B cells. We have identified several classes of small molecules that inhibit kinases that block the early signal transduction events controlling the activation of lymphocytes and other immune cells. These compounds are currently being evaluated in various models of immune disease.
Autoimmunity/Transplant Rejection Programs with Novartis
Regulate B cell activation. The goal of the B cell activation program is to prevent antibody secretion by activated B cells, an important mechanism in autoimmunity transplantation rejection. We have identified potential novel drug targets which we are currently validating. This program has been partnered with Novartis since August 1999. On May 15, 2002, Novartis notified us of its election to conclude the research phase of this collaboration on February 23, 2003.
Regulate T cell activation. The goal of our T cell activation program is to identify early steps in the process of T cell activation. T cells are responsible for cell-mediated inflammatory and humoral responses, both of which are important mechanisms of transplant rejection and autoimmune diseases. We have identified several novel drug targets in this program, of which one has been accepted by Novartis. The
23
program has been partnered with Novartis since May 1999. On May 15, 2002, Novartis notified us of its election to conclude the research phase of this collaboration on November 25, 2002.
Chronic Bronchitis
Chronic Bronchitis Program with Novartis – At Novartis Location
Epithelial cell activation. Using Rigel’s technology, Novartis is pursuing a program at its facilities for which the goal is to inhibit epithelial cell activation for the possible treatment of chronic bronchitis. This program is in the target screening and validation stage and may be terminated any time at Novartis’ discretion. Chronic bronchitis is a condition characterized by excessive mucus production that causes cough. It is associated with hyperplasia and hypertrophy of the mucus-producing glands found in the submucosa of large cartilaginous airways. Chronic bronchitis affects an estimated 5% of the U.S. population.
Cancer (Tumor Growth)
Cancer is a group of diseases characterized by the uncontrolled growth and proliferation of cells. This growth invades vital organs and often results in death. The United States market for branded cancer drugs totaled approximately $7.0 billion in 2001 and is projected to grow at an 11% annual growth rate. Cancer is the second leading cause of death in the United States, exceeded only by cardiovascular disease. In 2001, an estimated 1.3 million people were diagnosed with cancer, and more than 550,000 patients died of cancer in the United States. Although there have been improvements in cancer therapies over the last decade, there remains a significant medical need for the development of both more effective and less toxic drugs for these diseases.
Cancer Programs with Collaborators
Regulate cell cycle (Johnson & Johnson). This program is directed at finding new targets that regulate the cell cycle and the cell cycle checkpoint pathways. The proliferation of normal cells is controlled by built-in safety mechanisms in the cell cycle, termed checkpoints, that ensure that only cells with normal genetic material can progress through the cell cycle and divide. Cells with genetic mutations are recognized and shunted into the apoptosis pathway to protect the organism from cancer and other genetic disorders. It is estimated that more than 50% of all human tumors contain cancer cells that have lost one or more crucial checkpoint genes. Cancer cells also can carry mutations in another group of normal cell genes that mimic extracellular proliferation signals, causing tumor cells to continue to divide even in the absence of normal cell growth signals. The net result of these genetic mutations is uncontrolled cell division and disease. We have collaborated with our partner since December 1998 to identify intracellular drug targets involved in cell cycle control. We have identified several novel drug targets in this program, three of which have been accepted by Johnson & Johnson as validated. Two of these targets have completed high-throughput screening at Rigel and Johnson & Johnson with some of the identified compounds advancing to the lead profiling stage.
Inhibit tumor angiogenesis (Novartis). This antitumor program is directed toward the angiogenesis pathway. Angiogenesis is defined as the growth of new blood vessels. In diseased circumstances or in oxygen deficient conditions, angiogenesis is stimulated by the synthesis and release of specific pro-angiogenic factors. In contrast to normal angiogenesis, tumor angiogenesis is a continuous process. As a significant proportion of tumors are dependent on continued angiogenesis, inhibition of this process blocks tumor growth which often leads to complete tumor deterioration. Thus, we believe therapeutic intervention of tumor–promoted angiogenesis represents an important form of antitumor therapy. We have established and initiated multiple screens in human capillary endothelial cells and have identified several potential targets in the angiogenesis pathway. This drug discovery program for finding new targets for the development of small molecule inhibitors has been ongoing at Rigel for approximately two years and the collaboration with Novartis was initiated in July 2001.
Protein degradation oncology target (Daiichi). On August 1, 2002, we initiated a collaboration with Daiichi Pharmaceuticals to discover and develop drug candidates related to a specific protein degradation target. Under terms of the agreement, the initial stages of the collaboration will focus on the development of the assay for this specific target and the initiation of high-throughput screening to identify small molecules with oncology therapeutic applications.
Hepatitis C (Infectious Diseases)
Rigel-Owned Hepatitis C Programs
Inhibit viral replication. We acquired a cell based screening system in June 2001 to screen for small molecule inhibitors blocking the hepatitis C replication mechanism. We have completed compound screening and have identified several potential small molecule inhibitors of the hepatitis C replication mechanism. We are nearing the completion of the preclinical evaluation and plan to begin clinical testing in the second half of 2003.
Inhibit viral translation. We have initiated a viral research program based upon technology acquired from Questcor in September 2000. Hepatitis C is a major cause of chronic hepatitis, cirrhosis and hepatocellular carcinoma. The goal of this program is to interfere with the IRES translation mechanism of the hepatitis C virus. This program is currently in the compound screening stage of development.
24
Under the terms of our agreement with Questcor, we are obligated to assign back to Questcor all of our rights in the technology and intellectual property to which we are entitled pursuant to the agreement if we commit a material breach of the agreement and if Questcor follows certain procedures set forth in the agreement.
Ligase Initiative
The goal of the Ligase Initiative is to identify and determine the function of all ubiquitin ligases and ubiquitin proteases. These ligases and proteases are a very large family of enzymes that regulate the destruction of specific proteins in cells. Inappropriate activity of these enzymes has been implicated in cancer and autoimmunity, metabolic, cardiovascular and neurodegenerative diseases. It is believed that disease processes may be treated by either up-regulating or down-regulating these key signaling enzymes as a means of developing multiple therapeutic solutions.
Rigel-Owned Ligase Program for Tumor Growth and Inflammation
Control protein degradation (ligase). This program is focused on characterizing and developing specific inhibitors of protein-degrading enzymes, named ubiquitin ligases. Many intracellular proteins that play a critical role in signaling pathways are regulated by this protein-degrading process.
Disease processes can be treated by up-regulating or down-regulating these key signaling proteins as a way to enhance or dampen specific cellular responses. This principle has been successfully used in the design of a number of therapeutics for the treatment of inflammation. In the area of tumor growth, this program is focused on the ubiquitin ligase pathway unique to malignancies. The goal is to use specific inhibitors of ubiquitin ligases that regulate mitosis, or cell division, to stop growth and induce apotosis in transformed cancer cell lines. We continue to screen our library of small molecules against several members of the ubiquitin ligase family in order to identify small molecule compounds which may be potent and specific inhibitors.
Item 6. Exhibits and Reports on Form 8-K.
a) Exhibits:
The exhibits listed on the accompanying index to exhibits accompany or are filed or incorporated by reference (as stated therein) as part of this Quarterly Report on Form 10-Q.
b) Reports on Form 8-K:
None.
25
Pursuant to the requirements of the Securities Exchange Act of 1934, the registrant has duly caused this report to be signed on its behalf by the undersigned, thereunto duly authorized.
|
|
RIGEL PHARMACEUTICALS, INC. |
||
|
|
|
|
|
|
|
By: |
/s/ JAMES M. GOWER |
|
|
|
|
James M. Gower |
|
|
|
|
|
|
|
|
Date: |
November 14, 2002 |
|
|
|
|
|
|
|
|
By: |
/s/ JAMES H. WELCH |
|
|
|
|
James H. Welch |
|
|
|
Date: |
November 14, 2002 |
|
26
I, James M. Gower, Chief Executive Officer of Rigel Pharmaceuticals, Inc., certify that:
1. I have reviewed this quarterly report on Form 10-Q of Rigel Pharmaceuticals, Inc.;
2. Based on my knowledge, this quarterly report does not contain any untrue statement of a material fact or omit to state a material fact necessary to make the statements made, in light of the circumstances under which such statements were made, not misleading with respect to the period covered by this quarterly report;
3. Based on my knowledge, the financial statements, and other financial information included in this quarterly report, fairly present in all material respects the financial condition, results of operations and cash flows of the registrant as of, and for, the periods presented in this quarterly report;
4. The registrant’s other certifying officers and I are responsible for establishing and maintaining disclosure controls and procedures (as defined in Exchange Act Rules 13a-14 and 15d-14) for the registrant and we have:
a) designed such disclosure controls and procedures to ensure that material information relating to the registrant, including its consolidated subsidiaries, is made known to us by others within those entities, particularly during the period in which this quarterly report is being prepared;
b) evaluated the effectiveness of the registrant’s disclosure controls and procedures as of a date within 90 days prior to the filing date of this quarterly report (the “Evaluation Date”); and
c) presented in this quarterly report our conclusions about the effectiveness of the disclosure controls and procedures based on our evaluation as of the Evaluation Date;
5. The registrant’s other certifying officers and I have disclosed, based on our most recent evaluation, to the registrant’s auditors and the audit committee of registrant’s board of directors (or persons performing the equivalent function):
a) all significant deficiencies in the design or operation of internal controls which could adversely affect the registrant’s ability to record, process, summarize and report financial data and have identified for the registrant’s auditors any material weaknesses in internal controls; and
b) any fraud, whether or not material, that involves management or other employees who have a significant role in the registrant’s internal controls; and
6. The registrant’s other certifying officers and I have indicated in this quarterly report whether or not there were significant changes in internal controls or in other factors that could significantly affect internal controls subsequent to the date of our most recent evaluation, including any corrective actions with regard to significant deficiencies and material weaknesses.
Date: November 14, 2002
|
|
/s/ James M. Gower |
|
|
|
James M. Gower |
|
|
|
Chief Executive Officer |
|
27
CERTIFICATION
I, James H. Welch, Chief Financial Officer of Rigel Pharmaceuticals, Inc., certify that:
1. I have reviewed this quarterly report on Form 10-Q of Rigel Pharmaceuticals, Inc.;
2. Based on my knowledge, this quarterly report does not contain any untrue statement of a material fact or omit to state a material fact necessary to make the statements made, in light of the circumstances under which such statements were made, not misleading with respect to the period covered by this quarterly report;
3. Based on my knowledge, the financial statements, and other financial information included in this quarterly report, fairly present in all material respects the financial condition, results of operations and cash flows of the registrant as of, and for, the periods presented in this quarterly report;
4. The registrant’s other certifying officers and I are responsible for establishing and maintaining disclosure controls and procedures (as defined in Exchange Act Rules 13a-14 and 15d-14) for the registrant and we have:
a) designed such disclosure controls and procedures to ensure that material information relating to the registrant, including its consolidated subsidiaries, is made known to us by others within those entities, particularly during the period in which this quarterly report is being prepared;
b) evaluated the effectiveness of the registrant’s disclosure controls and procedures as of a date within 90 days prior to the filing date of this quarterly report (the “Evaluation Date”); and
c) presented in this quarterly report our conclusions about the effectiveness of the disclosure controls and procedures based on our evaluation as of the Evaluation Date;
5. The registrant’s other certifying officers and I have disclosed, based on our most recent evaluation, to the registrant’s auditors and the audit committee of registrant’s board of directors (or persons performing the equivalent function):
a) all significant deficiencies in the design or operation of internal controls which could adversely affect the registrant’s ability to record, process, summarize and report financial data and have identified for the registrant’s auditors any material weaknesses in internal controls; and
b) any fraud, whether or not material, that involves management or other employees who have a significant role in the registrant’s internal controls; and
6. The registrant’s other certifying officers and I have indicated in this quarterly report whether or not there were significant changes in internal controls or in other factors that could significantly affect internal controls subsequent to the date of our most recent evaluation, including any corrective actions with regard to significant deficiencies and material weaknesses.
|
Date: November 14, 2002 |
||
|
|
||
|
|
/s/ James H. Welch |
|
|
|
James H. Welch |
|
|
|
Chief Financial Officer |
|
28
INDEX TO EXHIBITS
|
Exhibit Number |
|
Description of Document |
|
3.1 |
|
Amended and Restated Certificate of Incorporation. (1) |
|
3.2 |
|
Amended and Restated Bylaws. (1) |
|
4.1 |
|
Specimen Common Stock Certificate. (1) |
|
4.9 |
|
Warrant issued to Comerica Bank – California for the purchase of shares of common stock. |
|
10.24 |
|
Loan and Security Agreement between Rigel and Comerica Bank - California, dated July 12, 2002. |
|
10.25 |
|
Collaboration Agreement between Rigel and Daiichi Pharmaceutical Co., Ltd., dated August 1, 2002. (2) |
|
15.1 |
|
Letter re unaudited interim financial information. |
|
99.1 |
|
Certification pursuant to Section 906 of the Sarbanes-Oxley Act of 2002. (3) |
(1) Filed with Rigel’s Registration Statement on Form S-1, as amended (No. 333-45864), and incorporated herein by reference.
(2) Confidential treatment requested as to specific portions, which portions are omitted and filed separately with the Securities and Exchange Commission.
(3) This certification accompanies this Quarterly Report on Form 10-Q and shall not be deemed “filed” by Rigel for purposes of Section 18 of the Securities Exchange Act of 1934, as amended.
29