safe and to help the communities where we live and work to reduce the number of people exposed to the virus. Through our existing Crisis Management Team (CMT), we implemented and continue to monitor our business continuity plans to prevent or minimize business disruption and ensure the safety and well-being of our personnel. Our CMT meets regularly to assess the effectiveness of our business continuity plans and make adjustments accordingly as COVID-19 continues to evolve. We have a COVID-19 Headquarters Policy (Plan) in place to provide guidelines when working onsite. We continue to evaluate the workplace for compliance with the local, state and federal guidance and may modify or update at any time to ensure the safety of our employees, contractors and visitors. During the first quarter of 2022, we updated our Plan as we move towards a hybrid schedule, reinstituting more in-person interactions back into our business beginning April 2022. We endeavor to provide the safest and most effective work environment under the circumstances, but we cannot guarantee that employees who come to the office will not be exposed to COVID-19 while at the office. It will be the responsibility of all employees to participate and cooperate in safety and cleaning protocols. We expect all employees, contractors, and visitors to our facility to comply with the Plan.
The ultimate impact of the COVID-19 pandemic on our business and financial condition is highly uncertain and subject to change, and as such, we cannot ascertain the full extent of the impacts on our sales of our products, our ability to continue to secure new collaborations and support existing collaboration efforts with our partners and our clinical and regulatory activities. Periodic resurgence of COVID-19 cases negatively impacted and may continue to impact our ability to grow our product sales. As COVID-19 cases surge, we have observed reduced patient-doctor interactions and our representatives are having fewer visits with health care providers. We continue to maintain our virtual engagements and as we see the declining trend in number of COVID-19 cases, we expect to continue to increase the in-person engagements with health care providers. We have plans in place to continue implementing both virtual and live initiatives to ensure we are able to meet the needs of health care providers as the pandemic continues to evolve.
With respect to our supply chain, we currently do not anticipate significant disruption in the supply chain for our commercial product. However, we do not know the full extent of the impact on our supply chain if the COVID-19 pandemic continues and persists for an extended period of time.
See also the section titled “Risk Factors” in Item 1A of this Form 10-Q for additional information on risks and uncertainties related to the ongoing COVID-19 pandemic.
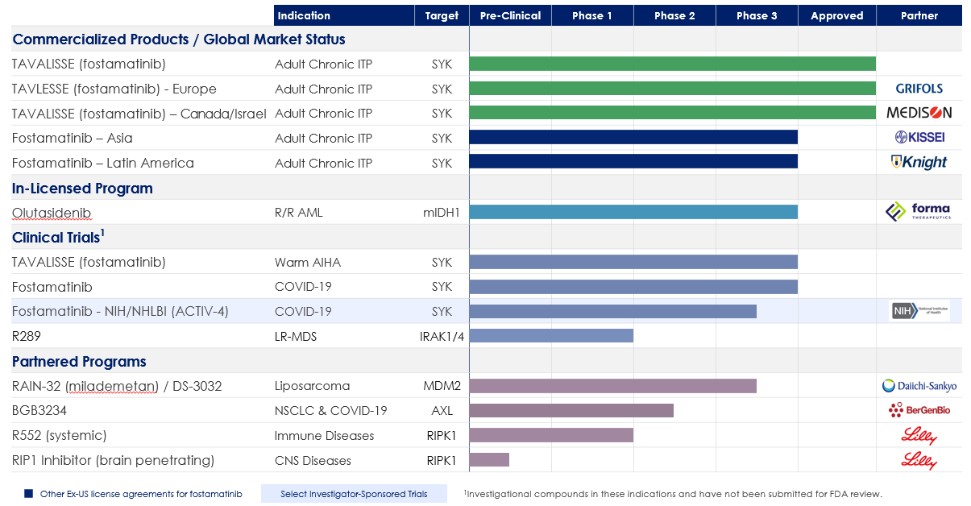
Our Product Portfolio
The following table summarizes our portfolio:
28